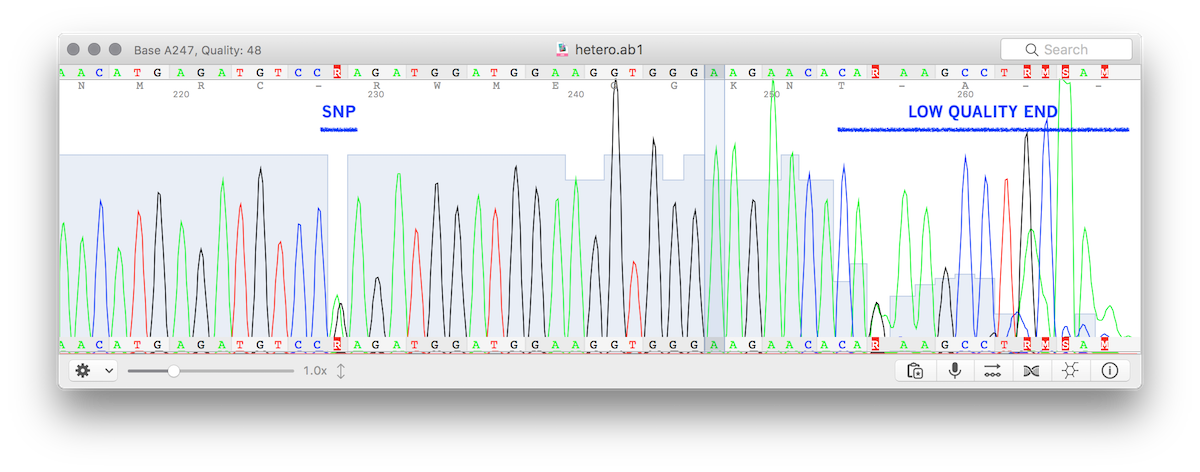
packageFASTX::Abi;use5.016;usewarnings;useBio::Trace::ABIF;useData::Dumper;useFile::Basename;$FASTX::Abi::VERSION='0.10.1';#ABSTRACT: Read Sanger trace file (chromatograms) in FASTQ format. For traces called with I<hetero> option, the ambiguities will be split into two sequences to allow usage from NGS tools that usually do not understand IUPAC ambiguities.our@valid_new_attributes= ('filename',# *REQUIRED* input trace filepath'trim_ends',# bool (default: 1) trim low quality ends'wnd',# int (default: 16) sliding window for quality trim'min_qual',# int (default: 22) threshold for low quality calls'bad_bases',# int (default: 2) maximum number of low quality bases per window'keep_abi',# bool (default: 0) import the Bio::Trace::ABIF object in FASTX::Abi (otherwise deleted after import));our@valid_obj_attributes= ('diff',# number of ambiguous bases'diff_array',# array of ambiguous bases position'sequence_name',# sequence name from filename'instrument',# Instrument'avg_peak_spacing',# Avg Peak Spacing in chromas'version',# version chromatograms'chromas',# Bio::Trace::ABIF object'hetero',# ambiguity'seq1',# Sequence 1 (non ambiguous, allele1)'seq2',# Sequence 2 (non ambiguous, allele2)'sequence',# Sequence, trimmed'quality',# Quality, trimmed'raw_sequence',# Raw sequence'raw_quality',# Raw quality'iso_seq',# Sequence are equal'discard',# Low quality sequence);our%iupac= ('R'=>'AG','Y'=>'CT','M'=>'CA','K'=>'TG','W'=>'TA','S'=>'CG');subnew {# Instantiate objectmy($class,$args) =@_;my$self= {filename=>$args->{filename},# Chromatogram file nametrim_ends=>$args->{trim_ends},# Trim low quality ends (bool)min_qual=>$args->{min_qual},# Minimum qualitywnd=>$args->{wnd},# Window for end trimmingbad_bases=>$args->{bad_bases},# Number of low qual bases per $window_widthkeep_abi=>$args->{keep_abi},# Do not destroy $self->{chromas} after use};#check valid inputs:formy$input(sortkeys%{$args} ) {if( !grep( /^$input$/,@valid_new_attributes) ) {confess("Method new() does not accept \"$input\" attribute. Valid attributes are:\n",join(', ',@valid_new_attributes));}}# CHECK INPUT FILE# -----------------------------------if(notdefined$self->{filename}) {confess("ABI file must be provided when creating new object");}if(not -e$self->{filename}) {confess("ABI file not found: ",$self->{filename});}my$abif;my$try=eval{$abif= Bio::Trace::ABIF->new();$abif->open_abif($self->{filename}) or confess"Error in file: ",$self->{filename};1;};if(not$try) {confess("Bio::Trace::ABIF was unable to read: ",$self->{filename});}my$object=bless$self,$class;$object->{chromas} =$abif;my@ext= ('.abi','.ab1','.ABI','.abI','.AB1','.ab');my($seqname) = basename($self->{filename},@ext);$object->{sequence_name} =$seqname;# DEFAULTS# -----------------------------------$object->{trim_ends} = 1unlessdefined$object->{trim_ends};$object->{wnd} = 10unlessdefined$object->{wnd};$object->{min_qual} = 20unlessdefined$object->{min_qual};$object->{bad_bases} = 4unlessdefined$object->{bad_bases};$object->{keep_abi} = 0unlessdefined$object->{keep_abi};$object->{discard} = 0;# GET SEQUENCE FROM AB1 FILE# -----------------------------------my$seq= _get_sequence($self);if($self->{keep_abi} == 0) {$self->{chromas} =undef;}#check valid attributes:formy$input(sortkeys%{$self} ) {# [this is a developer's safety net]# uncoverable condition falseif( !grep( /^$input$/,@valid_new_attributes,@valid_obj_attributes) ) {confess("Method new() does not accept \"$input\" attribute. Valid attributes are:\n",join(', ',@valid_new_attributes,@valid_obj_attributes));}}return$object;}subget_fastq {my($self,$name,$quality_value) =@_;if(notdefined$name) {$name=$self->{sequence_name};}elsif($name=~/\s+/) {$name=~s/\s+/_/g;}my$quality=$self->{quality};if(defined$quality_value) {if($quality_value=~/^\d+$/ and$quality_value>= 10) {my$q=chr(($quality_value<= 93 ?$quality_value: 93) + 33);$quality=$qxlength($quality);}elsif(length($quality_value) == 1) {$quality=$quality_valuexlength($quality);}else{confess("Supplied quality is neither a valid integer or a single char: <$quality_value>\n");}}my$output='';if($self->{iso_seq} ) {$output.='@'.$name."\n".$self->{seq1} ."\n+\n".$quality."\n";}else{$output.='@'.$name."_1\n".$self->{seq1} ."\n+\n".$quality."\n";$output.='@'.$name."_2\n".$self->{seq2} ."\n+\n".$quality."\n";}return$output;}subget_trace_info {my$self=shift;my$data;$data->{instrument} =$self->{instrument};$data->{version} =$self->{version};$data->{avg_peak_spacing} =$self->{avg_peak_spacing};return$data;}sub_get_sequence {my$self=shift;my$abif=$self->{chromas};$self->{raw_sequence} =$abif->sequence();# Get quality valuesmy@qv=$abif->quality_values();# Encode quality in FASTQ charsmy@fqv=map{chr(int(($_<=93?$_: 93)*4/6) + 33)}@qv;# FASTQmy$q=join('',@fqv);$self->{raw_quality} =$q;$self->{sequence} =$self->{raw_sequence};$self->{quality} =$self->{raw_quality};# Trimif($self->{trim_ends}) {#The Sequencing Analysis program determines the clear range of the sequence by trimming bases from the 5' to 3'#ends until fewer than 4 bases out of 20 have a quality value less than 20.#You can change these parameters by explicitly passing arguments to this method#(the default values are $window_width = 20, $bad_bases_threshold = 4, $quality_threshold = 20).# Note that Sequencing Analysis counts the bases starting from one, so you have to add one to the return values to get consistent results.my($begin_pos,$end_pos) =$abif->clear_range($self->{wnd},$self->{bad_bases},$self->{min_qual},);# This can be tested with low quality chromatograms# *TODO* to ask for some bad trace# uncoverable branch false# uncoverable condition left# uncoverable condition rightif($begin_pos>0 and$end_pos>0) {my$hi_qual_length=$end_pos-$begin_pos+1;$self->{sequence} =substr($self->{sequence},$begin_pos,$hi_qual_length);$self->{quality} =substr($self->{quality} ,$begin_pos,$hi_qual_length);}else{$self->{discard} = 1;}}# Check hetero basesif($self->{sequence}!~/[ACGT][RYMKWS]+[ACGT]/i) {$self->{hetero} = 0;}else{$self->{hetero} = 1;}# Check$self->{diff_array} = ();$self->{diff} = 0;my$seq1='';my$seq2='';for(my$i= 0;$i<length($self->{sequence});$i++) {my$q0=substr($self->{quality},$i, 1);my$s0=substr($self->{sequence},$i,1);# Ambiguity detected:if($iupac{$s0}) {my($base1,$base2) =split//,$iupac{$s0};$seq1.=$base1;$seq2.=$base2;$self->{diff}++;push(@{$self->{diff_array} },$i);}else{$seq1.=$s0;$seq2.=$s0;}}$self->{seq1} =$seq1;$self->{seq2} =$seq2;if($seq1eq$seq2) {$self->{iso_seq} = 1}else{$self->{iso_seq} = 0;}$self->{instrument} =$self->{chromas}->official_instrument_name();$self->{version} =$self->{chromas}->abif_version();$self->{avg_peak_spacing} =$self->{chromas}->avg_peak_spacing();}1;__END__=pod=encoding UTF-8=head1 NAMEFASTX::Abi - Read Sanger trace file (chromatograms) in FASTQ format. For traces called with I<hetero> option, the ambiguities will be split into two sequences to allow usage from NGS tools that usually do not understand IUPAC ambiguities.=head1 VERSIONversion 0.10.1=head1 SYNOPSISuse FASTX::Abi;my $trace_fastq = FASTX::Abi->new({ filename => '/path/to/trace.ab1' });# Print chromatogram as FASTQ (will print two sequences if there are ambiguities)print $trace_fastq->get_fastq();=head1 BUILD STATUS=for html <p><a href="https://travis-ci.org/telatin/FASTX-Abi" title="Test report"><img alt="TravisCI tests badge" src="https://travis-ci.org/telatin/FASTX-Abi.svg?branch=master"></a></p>The source from GitHub is tested using Travis-CI for continuous integration.Check also the CPAN grid test for a better estimate ofbuild success using CPAN version of interest.=head1 INSTALLATIONVia B<cpanminus>:# Install cpanminus if you don't have it:curl -L https://cpanmin.us | perl - --sudo App::cpanminus# Install FASTX::Abicpanm FASTX::AbiVia Miniconda L<https://docs.conda.io/en/latest/miniconda.html>:conda install -y -c bioconda perl-fastx-abi=head1 HETERO CALLING (IUPAC AMBIGUITIES)When Sanger-sequencing a mix of molecules (i.e. PCR product from heterozigous genome) containing a single-base polimorphisms,B<if> the I<.ab1> file is called using the I<hetero modality> the sequence stored in the file will contain ambiguous bases (i.e. using DNA IUPAC characters).This module is designed to produce NGS-compatible FASTQ, so when ambiguous bases are detected the two "alleles" will be split into two sequences(of course, if more SNPs are present in the same trace, the output I<cannot> be phased).The image below shows a trace file (.ab1) with a valid variant and a low quality end.=for HTML <p><img src="https://raw.githubusercontent.com/telatin/FASTX-Abi/master/img/chromatogram.png" alt="Sanger trace AB1" /></p>=head1 METHODS=head2 new()When creating a new object the only B<required> argument is I<filename>.# Trimming is based on Bio::Trace::ABIF->clear_range()my $trace_fastq = FASTX::Abi->new({filename => "$filepath",min_qual => 22,wnd => 16,bad_bases => 2,keep_abi => 1, # keep Bio::Trace::ABIF object in $self->{chromas} after use});# Raw sequence and quality:print "Raw seq/qual: ", $trace_fastq->{raw_sequence}, ", ", $trace_fastq->{raw_quality}, "\n";# Trimmed sequence and quality:print "Seq/qual: ", $trace_fastq->{sequence}, ", ", $trace_fastq->{quality}, "\n";# If there are ambiguities (hetero bases, IUPAC):if ($trace_fastq->{diff} > 0 ) {print "Differences: ", join(',', @{ $trace_fastq->{diffs} }), "\n";print "Seq 'A': ", $trace_fastq->{seq1}, "\n";print "Seq 'B': ", $trace_fastq->{seq2}, "\n";}Input parameters:=over 4=item I<filename>, pathName of the trace file (AB1 format)=item I<trim_ends>, boolTrim low quality ends (true by default, highly recommended)=item I<min_qual>, intMinimum quality value for trimming=item I<wnd>, intWindow size for end trimming=item I<bad_bases>, intMaximum number of bad bases per window=back=head2 B<get_fastq($sequence_name, $fixed_quality)>Return a string with the FASTQ formatted sequence (if no ambiguities) or twosequences (if at least one ambiguity is found).If no C<$sequence_name> is provided, the header will be made from the AB1 filename. If C<$sequence_name> is defined and contains spaces,they will converted to underscores.The C<$fixed_quality> is a user provided fixed quality value for each base printed. Can be an integer (10 < x < 40), or a single char.In the first case it will be encoded as quality score (values above 93 will all be rendered as C<~>), in the second case the characterwill be used as quality score. If not supplied the originalquality of the chromatogram will be used (that B<will be very low in SNPs positions>).# Use 40 as quality for each base of the trace:$trace->get_fastq(undef, 40);=head2 get_trace_info()Returns an object with trace information:my $info = FASTX::Abi->get_trace_info();print "Instrument: ", $info->{instrument}, "\n";print "Version: ", $info->{version}, "\n";print "Average peak distance: ", $info->{avg_peak_spacing}, "\n";=head2 _get_sequence()Internal routine (called by B<new()>) to populate sequence and quality.See new()=head1 SEE ALSOThis module is a wrapper around L<Bio::Trace::ABIF> by Nicola Vitacolonna.=head1 AUTHORAndrea Telatin <andrea@telatin.com>=head1 COPYRIGHT AND LICENSEThis software is Copyright (c) 2019 by Andrea Telatin.This is free software, licensed under:The MIT (X11) License=cut
{kind=link}
{kind=link}